
〒213-0002 神奈川県川崎市高津区二子1丁目7−17
リバーサイドマンション杉崎 102 二子新地駅 徒歩3分
| 月 | 火 | 水 | 木 | 金 | 土 | 日祝 | |
|---|---|---|---|---|---|---|---|
| 9:00〜13:00 | ● | ● | ● | ● | ● | ● | ─ |
| 15:00〜19:00 | ● | ● | ● | ● | ● | ● | ─ |
変形性関節症:炎症と軟骨破壊と痛みの負のループ
公開日:2026/06/18
更新日:2026/00/00
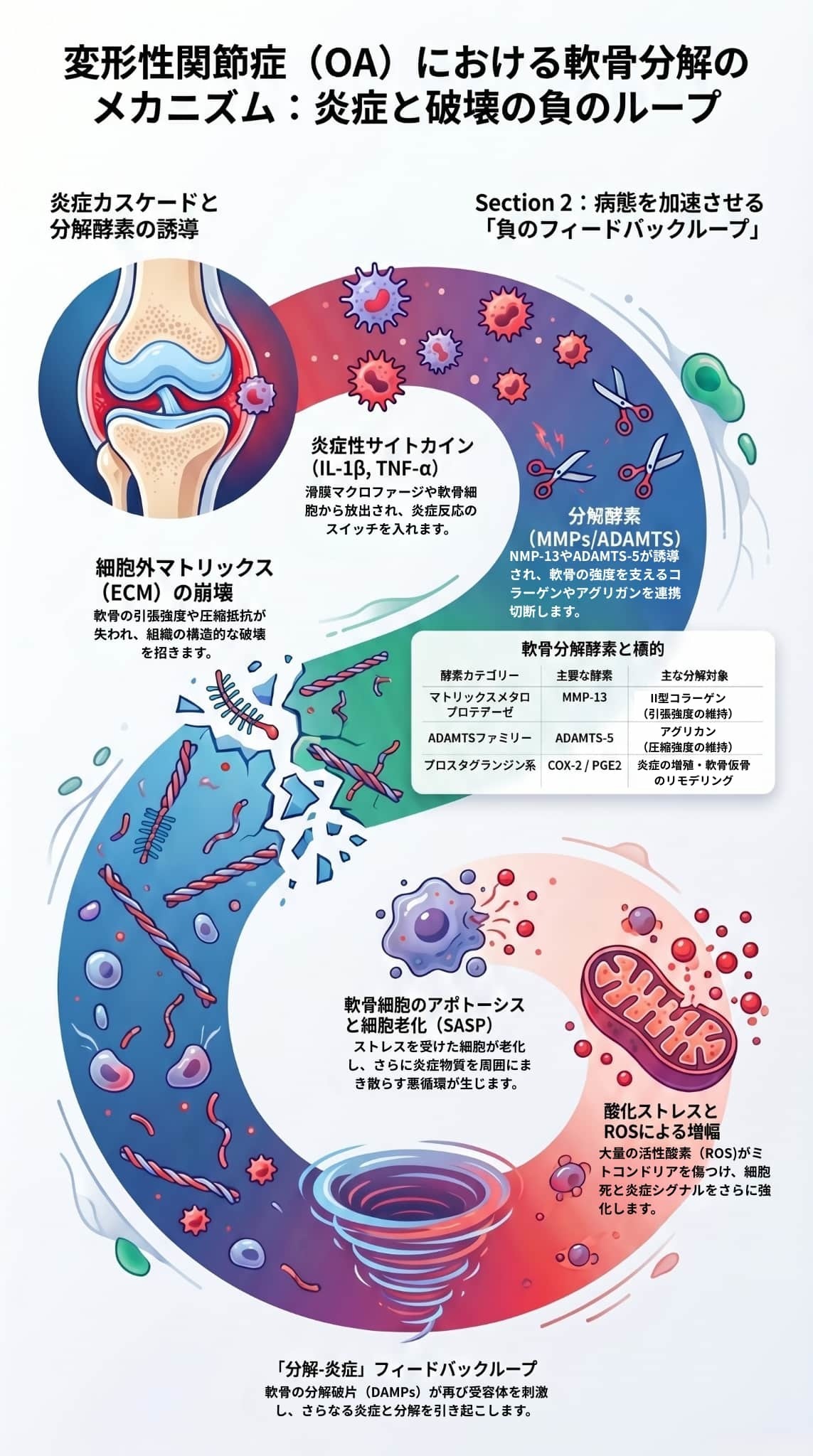
提供された資料は、変形性関節症(OA)における低悪性度の炎症が軟骨分解や関節痛を引き起こす複雑なメカニズムを学術的に概説しています。
疾患の本質を単なる摩耗ではなく、サイトカインや活性酸素種が関与する多因子的な器官障害として捉え、分子レベルでの病態を詳述しています。
軟骨細胞のアポトーシスや細胞老化、さらに軟骨下骨のリモデリングが、痛みの感作や組織破壊の悪循環を形成する様子が説明されています。また、現在の治療戦略の限界を指摘しつつ、セノリティクスやバイオマーカーを活用した個別化医療の必要性を提示しています。
最終的に、構造的変性と神経炎症の両面を標的とした包括的なアプローチが、次世代のOA管理において不可欠であると結論付けています。
目次
変形性関節症(OA)において、関節内の炎症物質が軟骨分解と関節痛を引き起こすプロセスは、関節全体の「低悪性度炎症(low-grade inflammation)」を起点とした複雑なネットワークによって進行します。
主要なメカニズムは以下の3つの段階に大別されます。
炎症性サイトカインは、軟骨の主要成分であるコラーゲンとプロテオグリカン(アグリカン)を直接破壊する分解酵素の産生を誘導します。
- MMP(マトリックスメタロプロテアーゼ)の誘導: IL-1βやTNF-αの刺激により、MMP-1、MMP-3、MMP-13などの分解酵素が発現します。中でもMMP-13は、軟骨の引張強度を保つ「II型コラーゲン」に対して最も高い分解活性を持ち、OA軟骨破壊の要となります。
- ADAMTSファミリーの誘導: 軟骨の圧縮抵抗性を担う「アグリカン」は、主にADAMTS-4およびADAMTS-5という酵素によって切断・分解されます。
- 悪循環(正のフィードバックループ)の形成: 破壊された軟骨断片はDAMPsとして働き、さらにマクロファージや軟骨細胞を刺激することで「分解-炎症」の悪循環を引き起こします。これに加えて、活性酸素種(ROS)による酸化ストレスや、老化・死滅した軟骨細胞がさらに炎症性因子をまき散らすこと(SASP:老化関連分泌表現型)で、分解が加速し続けます。
- 軟骨下骨の変化と神経侵入: 軟骨分解に伴い、軟骨の下にある骨(軟骨下骨)で骨吸収や骨髄病変(BMLs)が進行し、これが痛みの構造的基盤となります。病態が進行すると、軟骨下骨から軟骨へ感覚神経や血管が侵入し、痛みに直接寄与するようになります。
- 末梢性感作(痛覚過敏): IL-1β、TNF-α、IL-6などのサイトカインや、COX-2経路によって合成されるプロスタグランジンE2(PGE2)は、関節内の痛みを感知する神経(侵害受容器)を直接刺激します。これにより神経が過敏になり、通常は痛まないような無害な刺激でも痛みとして感じるようになります。
- 中枢性感作と神経ペプチドの関与: 炎症下では**神経成長因子(NGF)**の発現が増加し、痛みを伝える神経ペプチドである「サブスタンスP」や「CGRP」の合成が促進されます。これらの物質が放出されることで脊髄の神経回路が過興奮状態に陥り(中枢性感作)、関節の目立った損傷がなくても慢性的な強い痛みが持続するようになります。
変形性関節症(OA)は、関節軟骨の進行性変性を特徴とする慢性関節疾患であり、世界中で障害の主要な原因となっている疾患です。かつては加齢や負荷による単なる軟骨の「摩耗」と考えられていましたが、現在では関節軟骨だけでなく、軟骨下骨、滑膜、靭帯、半月板、神経要素を含めた関節全体を器官として侵す多因子性疾患として認識されています。病態としては、生体力学的因子と代謝因子の複雑な相互作用により、関節内の「低悪性度炎症(low-grade inflammation)」が引き起こされ、これが軟骨の分解や痛みの進行を駆動しています。
OAは世界的に非常に多くの人が罹患しており、有病率と発症率は年齢とともに増加します。
- 年齢と性別: 特に高齢者に多く、60歳以上では男性の10%、女性の18%が罹患しています。女性は男性よりも重症のOAを発症しやすく、とりわけ閉経後にその傾向が顕著になります。
- 罹患部位: 股関節よりも膝関節OAの有病率が高く、可動性を低下させる主要な筋骨格系疾患となっています。
- 将来の予測: 世界人口の25%以上が今後10年間で高齢化することから、OAの発症率は今後急激に上昇することが予測されています。
OAの発症と進行には、修正不可能な因子と修正可能な因子の両方が複雑に絡み合っています。
- 加齢: 発症の最大の危険因子であり、55歳以降になると軟骨分解の初期症状が現れ始めます。加齢に伴い、マトリックス合成や組織の修復能力が低下することが原因とされています。
- 肥満: BMIが30 kg/m²を超える人は膝関節OAに罹患しやすくなります。体重増加は関節への物理的負荷を増大させるだけでなく、軟骨分解の進行率を上昇させます(体重減少は痛みの改善に直結します)。
- 外傷(ケガ): 関節損傷は**全OA症例の約12%**を占めます。外傷後OAは、受傷から1年未満で早期に発症するケースもあれば、10〜20年もの間無症状のまま経過した後に発症するケースもあります。
- 遺伝的要因・生活習慣: COL2A1やADAMTS5といった遺伝的・エピジェネティックな異常のほか、運動不足、喫煙、多量のアルコール摂取などの不良な生活習慣も主要な危険因子として認識されています。
OAの進行により、患者は重度の関節痛やこわばり、著しい可動域の制限を経験します。これにより、日常の活動が制限されて患者個人の生活の質(QOL)や生産性が著しく低下します。さらに、世界的な人口の高齢化と肥満率の増加を背景に、長期的な患者管理が必要となるため、社会および公衆衛生サービスに対して重大な財政的・経済的負担をもたらす深刻な問題となっています。
変形性関節症(OA)における炎症カスケードは、関節内の「低悪性度炎症(low-grade inflammation)」の主要な病態である滑膜炎(synovitis)を中心とした複雑な分子機構によって駆動されます。このプロセスがどのように始まり、増幅していくのか、主要なステップをご説明します。
- フィブロネクチン断片: マクロファージのToll様受容体(TLRs)などを介して炎症反応を促進します。
- 低分子量ヒアルロン酸: TLR-4などを介して、マクロファージによる一酸化窒素(NO)や軟骨分解酵素(MMP)の産生を促します。
- HMGB1: 壊死した細胞などから放出され、サイトカインや分解酵素の産生を誘導します。
- 軟骨細胞: サイトカインやケモカインなどを自ら産生するようになり、同時にTLRsなどの受容体を発現して、細胞内の炎症・ストレス反応シグナルを活性化させます。
- 滑膜マクロファージ: 炎症反応と関節組織の破壊反応において「中心的な役割」を担う細胞です。実際に研究において、OAの滑膜細胞の培養系からマクロファージを特異的に取り除くと、主要な炎症性サイトカイン(IL-1βやTNF-α)の産生が消失し、その他の炎症因子(IL-6など)や軟骨分解酵素(MMP-1、MMP-3)も大幅に減少することが分かっています。
マクロファージや軟骨細胞の活性化によって、非常に強力な炎症性サイトカインであるIL-1βとTNF-αが滑液や滑膜、軟骨に放出されます。
- IL-1β: OA病態において最も強力な炎症性サイトカインの一つです。細胞内のNF-κBおよびMAPKシグナル伝達経路を活性化し、さらなる炎症因子(IL-6、IL-8)の放出や、軟骨を直接破壊する強力な分解酵素(MMPやADAMTS)の産生を誘導します。
- TNF-α: IL-1βと同様にNF-κB経路などを活性化し、分解酵素や痛みの原因となるプロスタグランジンE2(PGE2)の産生を刺激するとともに、軟骨マトリックスの合成を阻害します。
これらのサイトカイン(特にIL-1βとTNF-α)は相乗的に作用して滑膜炎を悪化させ、破壊的な炎症カスケードを急速に増幅させます。この滑膜炎の発生と増幅ループが起点となり、「軟骨細胞外マトリックス(ECM)の本格的な分解」と「慢性的な関節痛」の悪循環(分解-炎症の正のフィードバックループ)へと進行していきます。
変形性関節症(OA)における軟骨の分解は、滑膜炎などによって関節内に放出された強力な炎症性サイトカイン(特にIL-1βやTNF-α)が、軟骨の「細胞外マトリックス(ECM)」を直接破壊する分解酵素を過剰に誘導することによって進行します。
MMPsは、亜鉛に依存して軟骨マトリックスを分解する酵素のファミリーです。IL-1βやTNF-αの刺激によって軟骨細胞や滑膜マクロファージから産生されます。
- MMP-13(コラゲナーゼ-3): 軟骨の引張強度を保つ主要構造である**「II型コラーゲン」に対して最も高い分解活性を持ち、OAにおける軟骨破壊の要**となる非常に重要な酵素です。
- MMP-1(コラゲナーゼ-1): I型、II型、III型コラーゲンを切断して分解します。
- MMP-3(ストロメライシン-1): 広範な基質特異性を持ち、プロテオグリカンなどを分解するとともに、他のMMPsを活性化させる役割も担います。
軟骨に圧縮への抵抗性(クッション性)をもたらす主要なプロテオグリカンである「アグリカン」は、主にADAMTSファミリーという酵素群によって分解されます。 OA病態においては、ADAMTS-4とADAMTS-5が主要なアグリカナーゼ(アグリカン分解酵素)として機能し、これらもIL-1βやTNF-αなどの炎症性サイトカインによって誘導されます。
健常な軟骨では、MMPsによる「分解」と、それを抑制する内因性の組織阻害因子(TIMPs)による「保護」の間で動的なバランスが保たれています。 しかし炎症下のOA病態では、IL-1βなどの働きによってMMPsの発現が急増する一方で、TIMP-1やTIMP-2の発現が抑制されてしまうため、合成と分解のバランスが「分解(異化)優位」へと完全に傾いてしまいます。
これらの分解酵素の発現は、軟骨細胞内のNF-κB経路や**MAPK経路(ERK、JNK、p38など)といったシグナル伝達経路によってコントロールされています。一度軟骨の分解が始まると、以下のような要因によって破壊が自律的に加速する「分解-炎症」の正のフィードバックループ(悪循環)**が形成されます。
- 分解断片の刺激: 破壊されたアグリカンやII型コラーゲンの断片、あるいは損傷した細胞から出たdsRNAなどが細胞を直接刺激し、さらなる分解酵素の産生を促します。
- 酸化ストレスによる増幅: 活性酸素種(ROS)の過剰産生が、MMP-13などの発現を直接的に促進します。
- 細胞老化の影響: 分解や炎症のストレスで老化した軟骨細胞が、SASP(老化関連分泌表現型)と呼ばれる現象を引き起こし、自ら周囲にMMPsや炎症性因子を持続的にまき散らすようになります。
このように、複数の酵素の過剰産生、抑制因子の機能低下、そして分解産物自身が次なる炎症を呼ぶ悪循環が組み合わさることで、関節軟骨の不可逆的な喪失へと至ります。
変形性関節症(OA)において、活性酸素種(ROS)とそれがもたらす酸化ストレスは、炎症カスケードと軟骨の分解メカニズムをさらに増幅・加速させる非常に重要な役割を担っています。
これまでの滑膜炎や軟骨分解のメカニズムと密接に関わりながら、OA病態を悪化させる仕組みは以下のようにまとめられます。
- ROSの産生源: OA関節内では、軟骨細胞のミトコンドリアの機能障害や、NADPH酸化酵素、さらには活性化した炎症細胞(マクロファージなど)から大量の活性酸素種(ROS)が産生されます。
- 防御機能の崩壊: 健常な軟骨では、スーパーオキシドジスムターゼ(SOD)やカタラーゼ(CAT)といった抗酸化酵素が活性酸素種(ROS)のバランスを保っていますが、OA病態ではこの防御機構が破綻し、酸化ストレスが過剰な状態になります。
活性酸素種(ROS)は関節内の生体分子に直接的な損傷を与えます。 特に近年注目されているのが、酸化ストレスや代謝異常によって引き起こされる**「ジスルフィドトーシス(disulfidptosis)」**という細胞の崩壊プロセスです。NADPHの枯渇により細胞内に異常なジスルフィド結合が蓄積することで細胞が崩壊し、これがDAMPs(損傷関連分子パターン)やSASP(老化関連分泌表現型)因子を周囲に放出させ、さらなる炎症を引き起こす要因となります。
酸化ストレスは、軟骨を直接破壊する強力な酵素群の産生を後押しします。 活性酸素種(ROS)は、軟骨分解の要となるMMP-13などの発現を直接的に促進し、炎症性サイトカインと協力してII型コラーゲンやアグリカンの過剰な分解を駆動します。
活性酸素種(ROS)と、IL-1βやTNF-αといった炎症性サイトカインは、互いに相手の産生を促進し合う「正のフィードバックループ」を形成しています。 酸化ストレスが炎症細胞を刺激してサイトカインを出させ、そのサイトカインがまた細胞を刺激して活性酸素種(ROS)を産生させるという悪循環に陥ることで、軟骨の破壊とアポトーシス(細胞死)が止まらなくなります。
活性酸素種(ROS)と酸化ストレスは、軟骨細胞の代謝そのものも狂わせます。ミトコンドリア機能の低下は、グルコースの取り込み障害などの「代謝的ストレス」を引き起こし、これが軟骨変性の基盤となります。また、炎症下でのマクロファージの代謝変化(解糖系再プログラミング)などを引き起こし、炎症環境をさらに悪化させます。
このように、酸化ストレスは単なる「細胞のサビ」ではなく、**軟骨細胞を直接破壊し、分解酵素を活性化させ、さらには強力な炎症を持続させる「増幅器」**として、OAの進行を力強く推し進める病態の核心的なメカニズムの一つとして機能しています。
シクロオキシゲナーゼ(COX)経路は、変形性関節症(OA)における「炎症の増幅」と「痛みの発生」の両方を媒介する中心的な分子メカニズムです。この経路は、アラキドン酸から**プロスタグランジンE2(PGE2)**を生成し、これまで解説した炎症性サイトカインや軟骨分解酵素と密接に相互作用しながら病態を悪化させます。
主要なメカニズムは以下の通りです。
- EP4受容体: OA軟骨で発現が上昇しており、**PGE2が引き起こすマトリックス分解(MMP-13の誘導など)や、感覚神経の感作(痛覚過敏)を媒介する病態の「悪役」**として中心的な役割を果たします。
- EP2受容体: 反対に、EP2の刺激は軟骨細胞の増殖やアポトーシスからの保護など、組織再生を促進する保護的な作用を持つことが分かっています。
PGE2は軟骨を壊すだけでなく、神経系に直接作用して痛みを増幅させます。具体的には、後根神経節(DRG)のニューロンにおける特定のナトリウム電流(Nav1.8電流など)を増加させることで、痛みを感知する神経(侵害受容器)を過敏にします。
これにより、痛みのセンサーが通常より低い刺激で反応するようになる「末梢性感作」と、脊髄などの中枢神経系が過興奮状態になる「中枢性感作」の両方を強力に引き起こします。
現在のOA疼痛管理では、このCOX経路を阻害するNSAIDs(非ステロイド性抗炎症薬)が第一選択薬として広く使われています。しかし、単純にCOX阻害剤でこの経路だけをブロックすると、行き場を失ったアラキドン酸が別の炎症経路(5-リポキシゲナーゼ経路)へ流れ込み、ロイコトリエンB4(LTB4)という別の炎症物質の産生を3〜5倍に増やしてしまう現象(シャント)が確認されています。
変形性関節症(OA)において、これまでご説明してきた炎症性サイトカインや酸化ストレスなどが関節内に持続的に存在すると、軟骨細胞の運命を決定づける「アポトーシス(細胞死)」と「細胞老化」が引き起こされます。これらは軟骨の恒常性を崩壊させ、病態の進行を加速させる重要なプロセスです。
- アポトーシスの誘導: IL-1βやIL-6などの炎症性サイトカイン、過剰な活性酸素種(ROS)、そして異常な機械的ストレスが主要な引き金となります。これらのストレスが加わると、ミトコンドリア経路などを介して「カスパーゼ(特にカスパーゼ-3)」と呼ばれる酵素が活性化し、軟骨細胞を自死へと追いやります。
- パイロトーシス(炎症性細胞死)の関与: OA関節ではアポトーシスだけでなく、「パイロトーシス」と呼ばれるインフラマソーム(NLRP3など)によって誘発される激しい細胞死も発生します。細胞が膨張・破裂し、細胞内からIL-1βやIL-18などの炎症性サイトカインを周囲に大量に放出するため、さらなる炎症を引き起こします。
細胞老化は、OA病態プロセスの主要な駆動因子と考えられています。
- 老化のトリガー: 加齢に加え、炎症性サイトカイン、酸化ストレス、異常な機械的負荷、ミトコンドリアの機能不全(機械的・代謝的二重打撃)などが軟骨細胞の老化を促進します。
- SASP(老化関連分泌表現型)の脅威: 老化した軟骨細胞は単に活動を停止するだけではありません。「SASP」と呼ばれる現象により、自らIL-1β、IL-6、TNF-αといった炎症性サイトカインや、軟骨分解酵素(MMPsやADAMTS)を持続的に周囲へ分泌するようになります。
- 「分解-炎症」の悪循環の固定化: 老化細胞から撒き散らされたこれらの因子は、軟骨、滑液、軟骨下骨へと広がり、周囲の健康な細胞にまで二次的な損傷を与えます。これが新たな炎症と細胞の老化・死を呼び起こすという強力な悪循環を形成し、低悪性度炎症を持続させます。
アポトーシスや老化によって軟骨細胞の数が減少し、残った細胞もマトリックスを合成する能力を失うため、軟骨は自らを修復する能力を著しく損ないます。
近年、このメカニズムを逆手に取った**「セノリティクス(senolytic drugs)」**と呼ばれる新薬の可能性が注目されています。これは、関節内の「老化細胞」だけを選択的に除去する薬剤です。動物モデルの研究では、セノリティクスを投与することで痛覚過敏が軽減し、感覚神経への悪影響が抑えられることが示されており、OAの根本的な病態を修飾する新しい治療ターゲットとして期待されています。
変形性関節症(OA)は軟骨単独の病気ではなく、関節全体を巻き込む器官障害です。なかでも軟骨の下に位置する骨である**「軟骨下骨(subchondral bone)」の病的な変化は、軟骨の分解と並行して進行し、OAにおける「痛み」を決定づける極めて重要な役割を担っています**。
- 軟骨下骨から放出されるSDF-1という物質は、軟骨細胞(CXCR4受容体)に直接結合し、軟骨の変性を誘発します。
- また、IL-6などの因子は、軟骨下骨と軟骨のクロストークにおいて、軟骨の破壊(STAT3経路の活性化)を進めると同時に、神経の伸長や痛み神経の活性化(ERK経路の活性化)を促進するという「破壊と痛みの二刀流」の働きをしています。
参考文献に基づき、変形性関節症(OA)における「関節痛」のメカニズムについて解説します。
- 炎症性メディエーターの直接作用: IL-1βやTNF-αは、痛みを伝える神経線維(Aδ線維やC線維)を直接刺激し、機械的な刺激に対する感受性を高めます。また、プロスタグランジンE2(PGE2)は、後根神経節(DRG)のニューロンにおいて特定のナトリウム電流(Nav1.8電流など)を増加させることで、痛みのセンサーを強力に過敏にします。
- 構造的変化による神経侵入: OAの後期になると、軟骨下骨と軟骨の境界(潮汐線)を破り、軟骨下骨の側から軟骨へ感覚神経や交感神経を含む血管経路が侵入してきます。この異常な神経支配と、軟骨下骨に生じる骨髄病変(BMLs)や骨破壊が、痛みの発生と直接的に関連しています。
- 神経成長因子(NGF)の中核的な役割: NGFはOAの痛みにおいて中心的な役割を果たす因子です。滑膜などで産生されたNGFは、神経における痛みのセンサー(TRPV1チャネルやブラジキニン受容体)を増やすとともに、「サブスタンスP」や「CGRP(カルシトニン遺伝関連ペプチド)」といった痛みを伝える神経ペプチドの合成を増加させます。実際に、滑膜におけるNGFの発現量が高い患者ほど、中枢性感作のスコアが高いことが分かっています。
- 神経ペプチドによる脊髄の過興奮: 感作された神経から放出されたサブスタンスPやCGRPは、脊髄の過興奮性を直接的に促進し、痛みのシグナルを増幅させて中枢性感作を引き起こします。
- サイトカインの時期特異性: 興味深いことに、IL-1β、TNF-α、IL-6といった炎症性サイトカインの濃度は、OAの末期よりも「早期段階」で高く、これが早期の痛みの強さと強い相関を示します。
- ブラジキニン: 古典的な発痛物質であり、イオンチャネルを直接開いたり、神経の興奮性を変化させたりすることで痛みを誘導します。
変形性関節症(OA)の治療戦略は、これまでお話ししてきた「炎症カスケード」「軟骨分解」「細胞老化」「痛みの感作」といった複雑な病態メカニズムに基づいて開発が進められています。
現在、OAの痛みを管理するための第一選択薬として広く使われているのが**NSAIDs(非ステロイド性抗炎症薬)**です。
- これらは、痛みの強力な増幅器である「COX酵素」を阻害し、プロスタグランジンE2(PGE2)の産生を抑えることで痛みや腫れを軽減します。
- しかし、従来の非選択的NSAIDsは消化管への副作用リスクがあり、COX-2だけを特異的に阻害する薬は心血管系のリスクを高めるという課題があります。
- 近年では、COX経路と別の炎症経路(LOX経路)の両方を阻害する「リコフェロン」のような新薬が、臨床試験で軟骨喪失の減少や痛みの軽減を示しており、副作用の少ない治療法として期待されています。
軟骨分解の強力な引き金となるIL-1βやTNF-α、IL-6などの「炎症性サイトカイン」をブロックする生物学製剤の開発が盛んに行われてきました。しかし、現状ではこれらの多くが臨床試験で失敗に終わっています。
- 抗IL-1療法 / 抗TNF-α療法: カナキヌマブ(IL-1β標的)やアダリムマブ(TNF-α標的)などが試されましたが、ほとんどの試験で痛みの有意な改善や、軟骨喪失を食い止める効果(疾患修飾効果)を証明できませんでした。カナキヌマブは人工関節置換術のリスクを下げる傾向を示しましたが、重篤な感染症リスクが増加したため、全体的なメリットが相殺されてしまいました。
炎症そのものではなく、「痛みの神経メカニズム」を直接ブロックする戦略は有望な成果を上げています。
- 抗NGF療法: 中枢性感作の中心的な役割を果たす「神経成長因子(NGF)」を阻害する抗NGF抗体は、OAの痛みを「有意に軽減」することが確認されています。
- EP4受容体アンタゴニスト: PGE2が痛みを引き起こす際に結合する「EP4受容体」をブロックする薬(グラピプラントなど)は、すでにイヌのOA疼痛治療薬として承認されており、痛みの感作を防ぐ有効な手段となっています。
- セノリティクス薬: 関節内の「老化細胞」だけを選択的に除去する薬です。動物実験では、軟骨下骨からの痛み神経(CGRP陽性神経)の侵入を減らして痛みを軽減することが確認されましたが、病態進行中の骨や軟骨の構造的な変性自体は止めることができませんでした。
- 抗酸化療法(LXR活性化): 肝X受容体(LXR)を活性化させることで、サイトカインによる軟骨分解とPGE2産生を劇的に強力にブロックし、痛みを軽減するアプローチも前臨床段階で有望視されています。
- 時期特異性(タイミングの問題): IL-1βやTNF-αといった標的となる炎症性サイトカインは「OAの初期」に多く分泌され、「末期」には非常に低濃度になってしまいます。そのため、すでに病態が進行した患者にこれらの薬を投与しても効きにくいと考えられています。
- 患者の不均一性: 滑膜炎の重症度や代謝などの違いによって、痛みを引き起こしている主要な因子(NF-κBなのか、TNF-αなのかなど)が患者ごとに異なります。全員に同じ単一の標的薬を使っても効果が出にくいため、今後は患者ごとの「個別化医療」が必要とされています。
- 効果判定の難しさ: 軟骨分解は非常にゆっくり進行するため、新薬(DMOAD)の臨床試験は長期化しやすく、開発の大きな壁となっています。
膝変形性関節症(OA)の進行を根本から食い止める疾患修飾OA薬(DMOAD)は未だ実用化されておらず、サイトカイン標的療法の臨床試験も多くが失敗に終わっているのが現状です。これらを打ち破るための「将来の研究方向性と未解決の課題」は、主に以下の5つの領域に集約されます。
- 炎症の初期トリガーの特定: 炎症の引き金となるDAMPs(損傷関連分子パターン)が複数ある中で、疾患の早期段階において「どのDAMPsが最も重要なのか」、また「それらが時間的にどう相互作用するのか」が完全には分かっていません。
- 「画像所見と痛みの不一致」の謎: X線などの画像所見と患者が感じる痛みの強さが一致しない理由も大きな課題です。強い滑膜炎があるのに痛みが軽い患者がいる一方で、組織学的な炎症がないのに強い痛みを感じる患者がいる理由は未解明です。
- 軟骨修復の限界: 治療によって軟骨の厚みが回復したとしても、それが必ずしも臨床的な症状改善に結びつかない理由も解明が求められています。
- IL-33/ST2経路とLXRの活性化: 炎症を強力に促進するIL-33の遮断や、サイトカインによる軟骨分解と痛みの原因(PGE2)を劇的に抑え込む「肝X受容体(LXR)」の活性化が有望視されています。
- 組織特異的なシグナル阻害: IL-6の下流にあるJAK/STAT3(軟骨分解に関与)やERK(痛みの神経伸長に関与)を標的とする際、単に全体をブロックするのではなく「組織特異的」にコントロールする技術が必要です。
- エピジェネティクスと腸内細菌叢: miR-21-5pなどのエピジェネティクス制御のほか、腸内細菌叢(マイクロバイオーム)の異常がIL-17などを介して関節破壊に影響を及ぼす可能性も新たなターゲットとして浮上しています。
- 早期診断マーカー: 滑液中のIL-6や、軟骨マトリックスのリモデリングを示すC1Mなど。
- 高精度な血清パネル: TNF-α、IL-1β、IL-6などの血清マーカーを組み合わせて、OA患者を約97〜100%の精度で識別するパネル。
- 予後予測・治療反応性マーカー: IL-10やVEGFなど、将来の痛みや機能低下を予測するマーカーや、特定の薬が効くかどうかを判定するマーカー。
- 表現型に基づく層別化: NF-κB、TNF-α、TGF-βといった炎症マーカーの発現パターンや代謝性疾患の有無によって、OA患者を4つの異なるサブグループに分類し、それぞれに最適な治療を行うアプローチです。
- 性別・遺伝的要因の考慮: COL2A1などの遺伝的リスク評価や、「IL-6欠損による保護効果が雄マウスでのみ観察され、雌マウスでは観察されない」といった性特異的な病態への対応も重要になります。
[2]C. Coppola et al., “Osteoarthritis: Insights into Diagnosis, Pathophysiology, Therapeutic Avenues, and the Potential of Natural Extracts,” Current Issues in Molecular Biology, vol. 46, no. 5, pp. 4063–4105, Apr. 2024, doi: 10.3390/cimb46050251.
[66]L. Li, Z. Li, Y. Li, X. Hu, Y. Zhang, and P. Fan, “Profiling of inflammatory mediators in the synovial fluid related to pain in knee osteoarthritis.,” BMC Musculoskeletal Disorders, vol. 21, no. 1, pp. 99–99, Feb. 2020, doi: 10.1186/S12891-020-3120-0.
ごあいさつ

長引いた痛みを一人で治すのは困難なことが多いです。
困ったときは自身で判断せずに適切な処置を受けるために専門家に相談しましょう。
もし、お近くにお住まいで、困っているならば、一度ひまわり接骨院までお問い合わせください。腰痛・坐骨神経痛の専門家の新幡が、ご相談に乗ります。
気軽にご相談ください。

新着情報・お知らせ
お気軽にお問合せください

お電話でのお問合せ・相談予約
<受付時間>
月~土
9:00〜13:00 /15:00〜19:00
※日曜・祝日は除く
フォームは24時間受付中です。お気軽にご連絡ください。
- トップページ
ひまわり接骨院
住所
〒213-0002
神奈川県川崎市高津区二子1丁目7−17 リバーサイドマンション杉崎 102
アクセス
二子新地駅 徒歩3分
駐車場:近隣にコインパーキングあり。自転車・バイクは店舗前に駐輪場がございます。
受付時間
月~土
9:00〜13:00 /15:00〜19:00
定休日
日曜・祝日

